- Visibility 65 Views
- Downloads 25 Downloads
- DOI 10.18231/j.ijn.2022.053
-
CrossMark

Amyloid myopathy: A rare cause of quadreparesis with bulbar palsy
Introduction
Amyloidosis is a syndrome with diverse clinical manifestations rather than as a disease with a single etiology and uniform presentation. It is the result of the deposition of insoluble protein fibrils approximately 10 nm in diameter in a beta-pleated sheet configuration, disrupting normal tissue structure and function.
The differential diagnosis for an adult presenting with acquired proximal muscle weakness is broad and includes such entities as inflammatory myopathies, muscular dystrophies, drug-induced myopathy, Lambert- Eaton myasthenia syndrome, and endocrine myopathies, among others. Amyloid myopathy is a cause of proximal muscle weakness that is often overlooked because, although it is well characterized, it is rarely observed clinically.
Case Report
A 50 year old female with no previous co morbidities presented with progressive hip and shoulder muscle weakness of 1.5 years duration, followed by difficulty in swallowing with nasal regurgitation of food. She also had change in voice with nasal twang.
On examination, she had neck flexor weakness, bilateral facial muscle weakness with weak gag reflex. There was no ptosis or extra ocular weakness. She had bilateral, proximal more than distal; symmetrical both upper and lower limb weakness, with diminished reflexes and flexor plantar response. No wasting or pseudo hypertrophy of muscles. There was no sensory or cerebellar involvement.
On evaluation, inflammatory markers were normal, serum TSH, Serum Creatinine kinase was normal, connective tissue antibodies were negative, Serum Acetylcholine receptor antibodies were negative, Serum Immunoflorecense electrophoresis and cryoglobulin levels were negative. MRI of brain and cervical spine was normal. Nerve conduction studies showed reduced amplitude of motor action potentials of nerves in all 4 limbs and absent sensory action potential, Repetitive nerve conduction studies were normal, Electromyography showed low amplitude polyphasic waves with normal recruitment suggestive of myopathic changes.
Nerve biopsy showed no evidence of inflammatory cells infiltration, granuloma formation or vasculitis seen. Muscle biopsy suggested chronic Myopathic changes without inflammation. Abdominal fat pad biopsy-adipose tissue with extracellular pink acellular material at places. Thioflavin immunofloroscence staining was suggestive of amyloidosis.
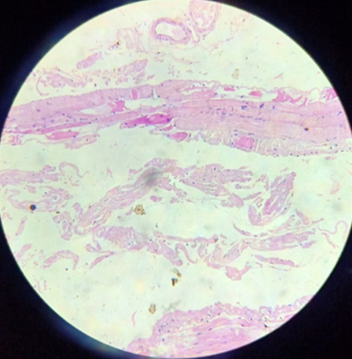
Discussion
Systemic amyloidosis may be acquired or hereditary. Acquired systemic amyloidosis may be primary (AL) or secondary (AA). AA amyloidosis occurs as a complication of chronic inflammation or infection, including rheumatoid arthritis, ankylosing spondylitis, Crohn disease, tuberculosis, osteomyelitis, and leprosy and most often affects the kidneys, liver, and spleen. Peripheral nervous system involvement is not a significant part of the clinical presentation of AA amyloidosis.[1]
Primary systemic amyloidosis (AL) is a rare disease. Its typical presentations are nephrotic syndrome, cardiomyopathy, peripheral neuropathy and hepatomegaly. One of the uncommon manifestation is amyloid myopathy (AM), the symptoms of which are usually non-specific, typically including progressive proximal weakness with an increased creatine kinase (CK) level, macroglossia and muscle pseudohypertrophy. Muscle weakness is atypical as the initial symptom of the disease.[2] AL amyloidosis and hereditary amyloidosis are both causes of neuromuscular dysfunction. Of the proteins responsible for systemic amyloidosis, only some are deposited in muscle causing myopathy. Dysphagia, weight loss, peripheral neuropathy and cardiomyopathy in patients with amyloid myopathy may serve as diagnostic clues for systemic amyloidosis. About 80% of patients with AL amyloid myopathy had no prior history of systemic amyloidosis or other plasma cell dyscrasias, and nearly 70% of patients with AL amyloid myopathy had myopathy as the sole initial manifestation at disease onset. [3]
The first recognized patient presenting with amyloid involvement in muscle was reported by Lubarsch in 1929. The term amyloid myopathy describes a clinically and etiologically heterogeneous group of myopathies, pathologically characterized by the presence of amyloid deposits in either the intramuscular blood vessels or connective tissue elements of the skeletal muscle. [4] The amyloid accumulation commonly involves multiple tissues, including muscle (systemic amyloidosis-associated myopathy), but occasionally it may be confined to the skeletal muscles (isolated amyloid myopathy). It is a rare myopathy even in patients with paraproteinemia and systemic amyloidosis. [3]
Patients with isolated amyloid myopathy have a younger age of onset compared to systemic amyloidosis (41.5 and 65 years). The symptoms of AM are usually non-specific, including progressive proximal weakness, macroglossia and muscle pseudohypertrophy as the typical features.Atrophy or normal muscle bulk may be found in majority of patients. Calf atrophy is only observed in patients with isolated amyloid myopathy and may serve as a diagnostic clue for the isolated amyloid myopathy group.
Thus differentiating from inflammatory myopathy may be difficult. Patients present with progressive muscular stiffness and weakness associated with muscle pseudo hypertrophy and macroglossia. Others present with atrophic myopathy without characteristic distinguishing features.
Serum creatine kinase levels may be mildly elevated, but often are normal as in our patients. MRI is useful in helping to target affected muscles for biopsy as well as for assessing disease activity. Metzler et al described 2 amyloid myopathy patients with the prominent finding of reticulation of the subcutaneous fat, coupled with negligible signal intensity alterations of the muscle.[3] The presence of monoclonal gammopathy usually is the key to identifying these patients as having amyloidosis.
Muscle biopsy is essential in the diagnostic evaluation of possible polymyositis, since metabolic abnormalities, muscular dystrophies, drug-induced changes, inclusion body myopathy, dermatomyositis, and amyloid myopathies. Spuler et al. found that the routine use of Congo red-stained sections increased the frequency of a diagnosis of AM 10-fold. [4] The pathologic diagnosis of polymyositis requires so-called “primary” endomysial inflammation by lymphocytes and macrophages surrounding and infiltrating non-necrotic myofibers. Necrosis, regeneration, and atrophy are also present but are nonspecific. It is important to note that the presence of several mononuclear cells solely in the perimysium does not constitute a diagnosis of polymyositis; indeed, this finding can be observed both in normal subjects and in patients with various myopathies.[5], [6] Our patient’s original H&E-stained slide sections showed atrophic changes in muscle fibres, Thioflavin immunofloresence staining showed accumulation of amyloid material. Hereditary amyloidosis can be caused by variants of transthyretin (TTR), apolipoprotein AI (apo AI), gelsolin, apolipoprotein AII, fibrinogen and lysosome. [1]
The treatment of Systemic amyloidosis depends on cardiac, renal, pulmonary status of individual, low risk patients receive autologous stem cell transplant with melphalan. Cyclophosphamide, Bortezomib and dexamethasone considered if bone marrow and plasma cell infiltration is more than 10%. The prognosis of amyloid myopathy is generally poor, with a mean time until death from onset of symptoms of 21.7 months (range of 2 weeks to 68.4 months). [7]
Thus, elderly patients who present with myopathy and dysphagia, weight loss with features suggesting inflammatory myopathy should prompt clinicians to consider a possibility of amyloid myopathy and request staining for amyloidosis, if it is not performed routinely, particularly in those whose biopsy findings are inconclusive.
Conflicts of Interest
All contributing authors declare no conflicts of interest.
Source of Funding
None.
References
- Z Simmons, CS Specht. The neuromuscular manifestations of amyloidosis. J Clin Neuromuscul Dis 2010. [Google Scholar] [Crossref]
- H Tuomaala, M Kärppä, H Tuominen, AM Remes. Amyloid myopathy: a diagnostic challenge. Neurol Int 2009. [Google Scholar] [Crossref]
- T Liewluck, M Milone. Characterization of isolated amyloid myopathy. Eur J Neurol 2017. [Google Scholar] [Crossref]
- S Spuler, A Emslie-Smith, A G Engel. Amyloid myopathy: an underdiagnosed entity. Ann Neurol 1998. [Google Scholar] [Crossref]
- Jp M, Jl F, Cl W, H Rg, Ep F, Rg G. MRI evaluation of amyloid myopathy. Skeletal Radiol 1992. [Google Scholar]
- KM Hull, L Griffith, RW Kuncl, FM Wigley. A deceptive case of amyloid myopathy: Clinical and magnetic resonance imaging features. Arthritis Rheum 2001. [Google Scholar] [Crossref]
- J E Chapin, M Kornfeld, A Harris. Amyloid myopathy: characteristic features of a still underdiagnosed disease. Muscle Nerve 2005. [Google Scholar] [Crossref]