Introduction
Hypothalamic Hamartoma (HH) are very rare tumor-like malformations that occur during fetal development and are present at birth. They are non-progressive lesions and do not spread or metastasize to other locations. The incidence is 1 in 200000 population.1 They will present in infancy and childhood with seizures, cognitive deficits,psychiatric symptoms, developmental delay and endocrinological manifestations. Rarely they may be asymptomatic also. We are presenting a case of large HH detected antenatelly and asymptomatic up to one year of age.
Case Report
A 14 month old girl presented with h/o spotting per vaginum 1 month back. She was first child of non consanguineous marriage with uneventful first trimester and second trimester. Routine antenatal USG at 29 weeks showed fetal brain hyperechoic round mass anterior to brainstem involving suprasellar area-displacing adjacent structures and Circle of Willis. Serial USG was showing mild increase in size of the lesion.
She was born induced at gestational age 36w+3d in view of severe preeclampsia. It was a late preterm low birth weight female baby (birth weight of 1.705 kg). Uneventful immediate postnatal period. No history of neonatal seizures or any other postnatal complications.
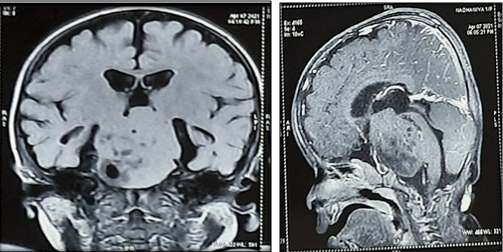
MRI taken at birth showed a well defined extra axial space occupying lesion with lobulated margins in suprasellar and pre-pontine cisterns measuring 4.1 X 3.9 X 3.6 cm with minimal post contrast enhancement and containing multiple serpiginous T2 hypointense components in the background of hyperintense stoma (Figure 1). Neurosurgery consultation was done and referred to higher center in view of size and site of lesion and low weight of baby. Discharged from our SNCU on postnatal day 11 with weight of 1.635kg. Came for follow up in SNCU OPD once and then lost follow up.
Now she presented with blood stained vaginal discharge at 13 months of age, lasted for 1 day. No history of trauma or drug intake. Baby is was having good appetite and sharing family pot diet. No increased frequency of urination noted. Child is was active and playful with good interaction with parents. No history of seizures- abnormal posturing, up rolling of eyes, weakness of limbs or history suggestive of cranial nerve palsies.
Developmental milestones were normal- corresponds to 10 months of age. From the weight records available with them her weight was below third centile at birth and it progressively increased and there was rapid increase after 6 month of age- reaching 90-97th centile now.
On examination child had stable vitals and head to foot examination unremarkable except for sexual maturity rating showing bilateral palpable breast tissue-B3 stage. Tanner staging was corresponding to B3A1P2. Weight length and head circumference was at 50-97th centile for age. System examination were within normal limits.
Blood routine investigations were normal. Hormonal study showed marked increase in LH & estrogen-LH: 5.38mIU/ml (normal <0.2mIU/ml) and Estrogen:282.7 pg/ml (normal 1-20 pg/ml). Her TFT, S.Cortisol and Prolactin levels were with in normal limits
Bone age was advanced corresponding to 24 months. USG breast confirmed presence of fibro glandular breast tissue. USG abdomen sshowed adult type uterus, corpus with thickened endometrium and prominent uterine corpus and ovarian cysts Pubertal-suggestive of pubertal morphology.
MRI brain at fourteen months of age Showed a fairly well defined extra axial lesion in the suprasellar cistern with mild interval increase in its size 4.97 X 4.9 X 3.5 cm - Suggestive of pedunculated large hypothalamic hamartoma presenting as precocious puberty[Figure 2].
She was treated with Inj Leuprolide acetate 3.75 mg IM monthly. We referred for early surgery but parents are not willing for the same.

Discussion
Hypothalamic hamartoma is a non-neoplastic malformation that appears in the hypothalamus. It is congenital, and it grows as the child’s brain grows, but HH doesn’t spread to other parts of the brain or body. The prevalence of this tumor in children and adolescents is approximately 1 case/200000. In 95% of cases, it is sporadic in origin, but rarely it may be an autosomal dominant disorder, when it is associated with Pallister-Hall syndrome.
HH present with diverse neurologic, endocrinologic, cognitive, behavioral and psychiatric comorbidities. 2 There are 2 types of clinical presentation of HH, intrahypothalamic and parahypothalamic, and each one is associated with a characteristic clinical spectrum. 3
Intrahypothalamic HH is associated with epilepsy. Gelastic epilepsy is very common in HH and is often pharmaco resistant. In any case of gelastic seizure one has to rule out possibility of HH.
Intra hypothalamic HH also manifest with developmental delay, cognitive deterioration, and psychiatric symptoms such as rage behaviors.3 Factors contributing to a greater degree of cognitive impairment and rage include higher number of anti-epilepsy medications taken, larger HH lesion size, younger age of seizure onset, and higher seizure frequency(3). About 40% of HH patients with epilepsy also have precocious puberty
Para hypothalamic HH is usually only associated with endocrinopathy, such as central precocious puberty (CPP). CPP due to HH will clinically manifest as early as 1-3 years of age. CPP is due to premature pulsatile release of gonadotropin-releasing hormone (GnRH) which in turn signals the ovaries or testes to begin production of sex hormones (estrogen and progesterone in females and testosterone in males).4 All cases of CPP should be investigated for HH. Rare endocrinological problems include acromegaly, diabetes insipidus, growth hormone deficiency, e19 central hypothyroidism, and the hypothalamic obesity syndrome. 5, 6
Surprisingly, irrespective of the large size of HH, some patients may have no symptoms at all.7 Our child also having very large HH, but asymptomatic till one year of age.
MRI is the investigation of the choice in HH. MR imaging on patients with epilepsy typically shows attachment of the HH in a posterior location in the hypothalamus, in the region of the mammillary bodies. MRI on patients with CPP shows attachment of the HH lesion in an anterior location in the hypothalamus, in the region of the tuber cinereum or pituitary stalk.8
Hormonal assay and other tests may be required according to the type of the HH and clinical presentation. EEG is useful in HH with seizure disorder, but it may be normal particularly in a case of gelastic seizures. This is due to the fact that gelastic seizures arise in structure located deep at the base of the brain, it is distant from EEG electrodes.9
Using whole exome sequencing, chromosomal microarray, somatic mutations of SHH pathway-related genes are discovered in one third of tissue and cell samples of patients with non-syndromic HH.10 Similarly, germline mutations in GLI3, is found in Pallister-Hall syndrome (AD), a form of syndromic HH.11
Advanced genotyping has shown that several other genes within the sonic hedgehog pathway can also have somatic mutations that result in HH. With current genotyping technology, somatic mutations can be identified in approximately 40% of HH lesions. But mutation analysis (genotyping) of HH lesions is not recommended for routine clinical care.10 Treatment include both medical and Surgical. Medical treatment is mainstay of treatment for children with seizures. Gelastic seizure is usually resistant to AEDs and may require surgery.12
CPP effectively treated with injection of gonadotropin-releasing hormone (GnRH) agonists, such as leuprolide acetate. Leuprolide will act by feedback inhibition of the pulsatile release of GnRH that is required to trigger puberty. Leuprolide acetate is administered as a once-monthly intramuscular injection for the duration of time that puberty needs to be suppressed.6
Several surgical approaches like microsurgical resection or disconnection, endoscopic resection have been proposed for resecting hamartomas.13 However, all those procedures entail substantial surgical risks. Several unconventional surgical procedures with good outcomes have been developed recently.14 It include gamma-knife radiosurgery, radio active seed implants, radio frequency ablation etc.15
The characteristic features our case include antenatal detection of a large HH and its asymptomatic course up to one year of age.