- Visibility 316 Views
- Downloads 49 Downloads
- Permissions
- DOI 10.18231/j.ijn.2021.053
-
CrossMark

Abstract
Motor neuron diseases are a group of chronic sporadic and hereditary neurological disorders characterized by progressive degeneration of motor neurons. These might affect the upper motor neurons, lower motor neurons, or both. The prognosis of the motor neuron disease depends upon the age at onset and the area of the central nervous system affected. Amyotrophic lateral sclerosis (ALS) has been documented to be fatal within three years of onset. This activity focuses on amyotrophic lateral sclerosis as the prototype of MND, which affects both the upper and the lower motor neurons and discusses the role of inter-professional team in the differential diagnosis, evaluation, treatment, and prognostication. It also discusses various other phenotypes of MND with an emphasis on their distinguishing features in requisite detail.
Introduction
Motor neuron diseases area unit a bunch of diseases that lead to progressive destruction of neurons and gradual deterioration of striated muscle function, resulting in high mortality and morbidity. The average incidence of MNDs is 1–3 cases per 100, 000.[1] The prevalence of MND ranges from one to nine cases per 100,000 worldwide. Of this cluster, amyotrophic lateral pathology is far and away the foremost common, comprising roughly 80%–90% of MND cases. [2] However, studies have shown a minimum of a 10% error in diagnosis of ALS. ALS and MND, in general, present with nonspecific symptoms like limb weakness, fasciculation’s, and fatigue, which might comprise each classic upper/bulbar and lower nerve cell clinical symptoms. Additionally, there aren't any reliable markers to differentiate ALS from alternative styles of MND, leading to diagnostic dilemmas. [3], [4]
In the United Kingdom, the term "motor neuron disease" (MND) is more commonly used. MND is a disease that strikes people in their middle to late years of life, with an average onset age of 58 years.[5] Despite being the third most prevalent neurodegenerative illness after Alzheimer's and Parkinson's, MND is rather rare, with an apparent uniform incidence of about 2/100,000 in areas where epidemiological data is available. [6] Despite its rarity, the disease has sparked a lot of interest because of its terrible course, which has put it at the centre of the ethical discussion over end-of-life decision-making and physician-assisted suicide. [7]
The incidence of MND is claimed to be growing, however this is most likely due to better diagnosis, improve knowledge of the disease, and an ageing population.[8] The incidence rises after the age of 40, peaks in the late 60s and early 70s, and then rapidly drops. The majority of patients die within two to five years of being diagnosed. According to recent research, the male to female ratio in MND is approaching one. 5–10% of the instances are familial, with the remainder being random. [9]
Etiology
In MND, higher motor neurons in the motor cortex or brainstem and spinal cord are degenerating, or both are degenerating, according to the clinical spectrum. MND has several variants, but the most prevalent is amyotrophic lateral sclerosis (ALS), which causes upper and lower motor neuron signs and symptoms. In one study, 94 percent of patients said they were satisfied with their treatment. [10], [11] In one study, 94% of patients had the ALS variation of MND. The appearance of upper and lower engine neuron includes as a rule starts centrally, and advances to include coterminous locales of the body with diminishing severity. Approximately 66% of cases start in the appendages and 33%, in the bulbar gathering of muscles; just a tiny level of them start with respiratory muscle involvement. [12]
There square measure 2 alternative main variants of MND: primary lateral pathology (PLS) and progressive muscular atrophy (PMA). One study showed that PLS accounts for around two of MND cases, and PMA for four-dimensional of cases. Several of those patients tend to accomplish the ALS variant over time. [13] Progressive neural structure palsy could be a term that's usually accustomed describe the neural structure onset of MND. [14]
Compared to patients with Associate in Nursing ALS variant, patients with PLS tend to gift 5 to 10 years earlier, have less limb wasting and neural structure symptoms throughout the course of the illness, and survive six to seven years longer. [15] The PMA is also associated is related to a degeneration of lower motor neurons of the medulla spinals within the absence of upper motor neurons. Cognitive impairment is highly identified in MND. Various subclinical cognitive defect and frontal lobe dysfunction may be responsible for MND.[16] Various genetic mutations are associated with MND like as TAR DNA-binding (TDP-43), which is responsible for front temporal dementia or Parkinson etc. [17]
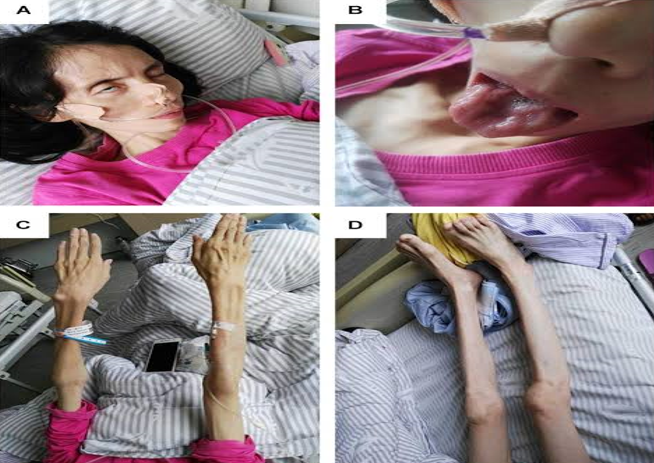
Epidemiology
The incidence of motor neuron disease has been shown to approach 2 to 3 per 100,000 populations, while a lower frequency (less than 1 per 100,000) of ALS has been demonstrated in the South and East Asian community. [18]
Ancestral origin has been shown to have a significant impact on the disease risk in ALS, with higher survival in racially heterogeneous or admixed populations (as compared to the White or Black race community). A shorter survival has also been reported in European ALS patients (2 years) as compared to the Asian population (4 years). [19]
Approximately 10 to 15 percent of individuals with ALS have familial disease. The estimated lifetime risk of sporadic ALS is one in 400. While bulbar onset ALS has been shown to be more common in females, spinal onset illness has been shown to be common in males. Progressive muscular atrophy (PMA) represents 2.5 to 11 percent of cases with MND. With an incidence of 0.02 per 100,000 populations, it is a much rarer form of the disease. It is predominantly seen in males, with a male to female ratio ranging from 3 to 7.5 to 1. The median age of onset is of 68 years, which is older than that of ALS patients. [20]
Differential Diagnosis
The most common disease that mimics the amyotrophic lateral sclerosis is none other than degenerative spondylotic myeloradiculopathy in the cervical and/or lumbosacral spine, which may present with radicular pain. This may be characterized by progressive symptoms that plateau later with the progression of the disease. Although imaging has been advised to differentiate between the two entities, there may be the presence of co-existent degenerative illness in those with ALS.[21]
Complications
Most patients with ALS die of respiratory failure within three years of the onset of the disease. Progressive weakness and wasting of the limb and respiratory muscles have been attributed to be the underlying precipitants of respiratory failure. Severe dysphagia may lead to weight loss, choking, and aspiration. Prolonged effortless mealtimes and coughing on attempting to swallow are other factors that have the ability to impact the patient's quality of care. [22]
Progressive deterioration in the activities of daily living, inability to ambulate, issues related to prolonged immobilization such as superficial skin infections, deceits ulcers, and deep venous thrombosis are also seen in these patients.[23]
Enhancing Healthcare Team Outcomes
A multidisciplinary team consisting of neurologists, respiratory physicians, physical medicine, and rehabilitation specialists (respiratory physiotherapists), gastroenterologists, dieticians may have an active role in the management of the patient. Palliative medicine input may be required for the management of pain, difficult to control symptoms (such as resistant breathlessness at the end of life), and psychosocial issues.[24] The inclusion of social care practitioners and staff who have been trained to manage patients, in the setting of their homes, in the multidisciplinary team has also been advised. Identification of the unique needs of family and provision of coordinated care in the home setting may become necessary as the patient approaches the terminal stage of decline. Good end of life care may be provided by specialist palliative teams. [25].
Acknowledgment
We owe our gratitude to all those researchers who have made this review possible. This review did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The author declares no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Source of Funding
None.
References
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344(22):1688-700. [Google Scholar] [Crossref]
- VY, Patil A, Shah P. Amyotrophic Lateral Sclerosis and its Mimics/Variants: A Comprehensive Review. J Clin Imaging Sci. 2018;8. [Google Scholar] [Crossref]
- Román GC. Neuroepidemiology of amyotrophic lateral sclerosis: Clues to aetiology and pathogenesis. J Neurol Neurosurg Psychiatry. 1996;61(2):131-7. [Google Scholar] [Crossref]
- Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: A population‑based study. Arch Neurol. 2000;57(1):109-13. [Google Scholar] [Crossref]
- Ringel SP, Murphy JR, Alderson MK. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-22. [Google Scholar] [Crossref]
- Traynor BJ, Codd MB, Corr B. Incidence and prevalence of ALS in Ireland, 1995-1997: a population-based study. Neurology. 1999;52(3):504-9. [Google Scholar] [Crossref]
- . Aosta register for ALS. Incidence of ALS in Italy: evidence for a uniform frequency in western countries. Neurology. 2001;56(2):239-44. [Google Scholar] [Crossref]
- Leigh PN, Ray-Chaudhuri K. Motor neuron disease. J Neurol Neurosurg Psychiatry. 1994;57(8):886-96. [Google Scholar] [Crossref]
- Logroscino G, Traynor BJ, Hardiman O. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry. 2008;79(1):6-11. [Google Scholar] [Crossref]
- Sathasivam S. Motor neurone disease: clinical features, diagnosis, diagnostic pitfalls and prognostic markers. Singapore Med J. 2010;51(5):367-72. [Google Scholar]
- Norris F, Shepherd R, DE. Onset, natural history and outcome in idiopathic adult onset motor neuron disease. J Neurol Sci. 1993;118(1):48-55. [Google Scholar] [Crossref]
- Norris F, Shepherd R, DE. Onset, natural history and outcome in idiopathic adult onset motor neuron disease. J Neurol Sci. 1993;118(1):48-55. [Google Scholar] [Crossref]
- Gordon PH, Cheng B, Katz IB. The natural history of primary lateral sclerosis. Neurology. 2006;66(5):647-53. [Google Scholar] [Crossref]
- Tartaglia MC, Rowe A, Findlater K. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch Neurol. 2007;64(2):232-6. [Google Scholar] [Crossref]
- Takeda S, Yamada M, Kawasaki K. Motor neuron disease with multi-system involvement presenting as tetraparesis, ophthalmoplegia and sensori-autonomic dysfunction. Acta Neuropathol. 1994;88(3):193-200. [Google Scholar] [Crossref]
- Ringholz GM, Appel SH, Bradshaw M. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586-90. [Google Scholar] [Crossref]
- Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008;70(2):144-52. [Google Scholar] [Crossref]
- Alonso A, Logroscino G, Jick SS. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol. 2009;16(6):745-51. [Google Scholar]
- Gundogdu B, Al-Lahham T. Racial differences in motor neuron disease. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(1-2):114-8. [Google Scholar]
- Liewluck T, Saperstein DS. Progressive Muscular Atrophy. Neurol Clin. 2015;33(4):761-73. [Google Scholar]
- Williams TL. Motor neurone disease: diagnostic pitfalls. Clin Med (Lond). 2013;13(1):97-100. [Google Scholar]
- Jackson CE, Mcvey AL, Rudnicki S, Dimachkie MM, Barohn RJ. Symptom Management and End-of-Life Care in Amyotrophic Lateral Sclerosis. Neurol Clin. 2015;33(4):889-908. [Google Scholar]
- Chen JH, Wu SC, Chen HJ, Kao CH, Tseng CH, Tsai CH. Risk of developing pressure sore in amyotrophic lateral sclerosis patients - a nationwide cohort study. J Eur Acad Dermatol Venereol. 2018;32(9):1589-96. [Google Scholar]
- Karam CY, Paganoni S, Joyce N, Carter GT, Bedlack R. Palliative Care Issues in Amyotrophic Lateral Sclerosis: An Evidenced-Based Review. Am J Hosp Palliat Care. 2016;33(1):84-92. [Google Scholar]
- Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017;10:205-15. [Google Scholar] [Crossref]