Introduction
The brain is one of the most advanced and superior structure in the human body. It is created from neurons and neuroglia, the neurons being responsible for sending and receiving nerve impulses or signals. The microglia and astrocytes are essential for ensuring applicable functioning of neurons. They may be quick to interfere while neurons become injured or stressed. As they're sentinels of neuron properly, pathological impairment of microglia or astrocytes also have devastating result for brain function. Acute injury causes neurons to come up with signals that inform neuroglia concerning the neuronal status. Relying on how severe a diploma of neuronal damage, neuroglia will each contend with the injured neurons into regeneration or kill them if they are not possible.1
The degeneration of CNS is characterized through chronic revolutionary loss of the shape and features of neuronal materials, consequent in persistent and mental impairments. Neurodegenerative diseases are characterised by the lack of neurons and modern dysfunction main to the first rate involvement of sensible systems defining scientific displays. The pathological additives involve the permeability of the blood brain barrier, the harm of myelin sheath, axon injury, the glial scar formation and the incidence of inflammatory cells, usually lymphocytes are infiltrated into the CNS. The lack of myelin is manifested in scientific symptoms together with neuropathic pain, paralysis, muscle spasms and optic neuritis.2
The process of neurodegeneration exists in intracellular procedures which incorporate apoptosis, autophagy, mitochondrial feature, oxidative strain, proteasome. Pathways related to vicinity of tissue surroundings (cell adhesion, endocytosis, neurotransmission, prions or transmissible elements), pathways related to systemic surroundings (infection/immune system, lipid/metabolic/endocrine, vascular elements), and techniques applicable to development and ageing (epigenetics, neurotrophic factors, telomeres).3
Neurodegenerative ailment known as degenerative myelopathy, which may be brought on thru through mutations in the same two genes that is frequently mutated in human ALS (amyotrophic lateral sclerosis) superoxide dismutase 1 (SOD1). The statement that cannabinoid receptor type-2 (CB2) expression is upregulated in human ALS.4
In this standpoint series, selected molecular advances appropriate to the biology of neurodegeneration process such as apoptosis, oxidative stress, and mitochondrial dysfunction will be reviewed.5 In this review, we systematically collected the molecular information probing the mechanisms of neurodegeneration.
Mechanism of Neurodegeneration
The prevalence of neurodegenerative diseases, inclusive of Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Frontotemporal dementia (FTD), and Huntington’s disease (HD).6
Neurotoxins constitute specific chemical mechanism, supply a way to
Advantage insight into cell mechanisms of apoptosis and necrosis,
Acquire a morphological template for studies otherwise impossible,
Specifically produce a unique phenotype of denervation, and
Provide the starting point to delve into processes and mechanisms of nerve regeneration and sprouting.21,22
Neurodegeneration includes
Unknown signaling cascades,
Misfolded proteins,
Protofibril formation,
Ubiquitin-proteasome dysfunction,
Oxidative and nitrosative stress, mitrochondrial injury.
Many greater activities which includes N-methyl-Daspartic acid (NMDA) receptors, voltage gated calcium channels (VGCCs), neuronal nitric oxide synthase (nNOS), oxidative stress from reactive oxygen species (ROS), and protein aggregation.7
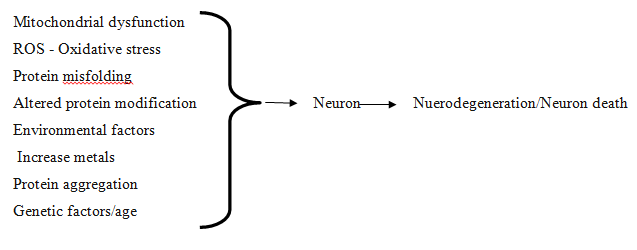
There is a big frame of evidence that advise these problems arise through manner of multifactorial situations including (Fig 1)
Ordinary protein dynamics with broken protein degradation and aggregation
Oxidative stress and free radical formation
Impaired bioenergetics and mitochondrial disorder,
Exposure to metal toxicity and pesticides.8
Free Radicals
Free radicals are consist of at least one unpaired electron in the outermost shell, is fairle reactive. The most common advised cellular free radicals are
Hydroxyl (OH•),
Superoxide (O2•),
Nitric oxide (NO•),
Nitrogen dioxide (NO2•),
Peroxyl (ROO•) and
Lipid peroxyl (LOO•).
Molecules such as hydrogen peroxide (H2O2), ozone (O3), singlet oxygen (1O2), hypochlorous acid (HOCl), nitrous acid (HNO2), peroxynitrite (ONOO-), dinitrogen trioxide (N2O3), lipid peroxide (LOOH), even as no longer appeared free radicals, can results easily cause free radical reactions in living organisms. Cells exposed to environment fortified with oxygen always generate oxygen free radicals. ROS includes oxygen-associated free radicals and reactive species, and they may be formed as a result of cardio metabolism.9
Oxygen is the mediator of electron–delivery mechanism, a dysfunction in the electron–shipping mechanism is suggestive of disrupted homeostatic regulation. And lots of experimental and clinical research assist a most vital disruption in homeostatic system in neurodegenerative situations. Respiratory chain deficits, mitochondria dysfunction, harmful reactive oxygen species developed, low hemoglobin levels, deficiency within the terminal complicated of the mitochondrial electron transport chain-cytochrome c oxidase (COX), defects in oxidative phosphorylation (Oxphos) has been observed within the pathophysiology of neurodegenerative conditions.10
Formation of ROS can appear in 2 ways that is enzymatic and non-enzymatic reactions. Enzymatic reactions producing free radicals include of those involved in the mitochondrial breathing chain, phagocytosis, prostaglandin synthesis and the cytochrome P450 system.9 As an instance, the superoxide radical is generated through a numerous cellular oxidase systems consisting of 5,10-methylenetetrahydrofolate reductase oxidase, xanthine oxidase, peroxidases. ROS can also be comprised of non-enzymatic reactions of oxygen with natural compounds in addition to those initiated by using ionizing radiations.
The non-enzymatic process also can appear at some point of oxidative phosphorylation in the mitochondria. ROS is generated from each endogenous and exogenous sources.
Endogenous free radicals are generated from immune cell activation, inflammation, mental stress, excessive exercise, ischemia, infection, most cancers and aging.
The impact of exogenous ROS from cigarette smoke, alcohol, air pollution, water pollution, heavy or transition metals (Cd, Hg, Pb, Fe, As), certain medicines (cyclosporine, tacrolimus, gentamycin, bleomycin), commercial solvents, cooking (smoked meat, used oil, fat) and radiation. After penetrated into the body by via precise routes, these exogenous compounds are decomposed or metabolized into free radicals. Generation of ROS can take place in a number organelles, mitochondria is the primary deliver of ROS production. The period of mitochondrial ROS is a final end result of oxidative phosphorylation, a technique that takes place in the inner mitochondrial membrane and entails the oxidation of reduced structure of nicotinamide-adenine dinucleotide to produce energy.9
Generation of ros in brain
Cellular ROS are generated typically through the use of at the same time exogenous and endogenous assets. Exogenous sources of ROS technology include ultra violet (UV), ionizing radiation, drug treatments whose mechanism of action is mediated by way of manner of ROS production. Environmental pollutions and chemical substances can also produce ROS as a follow-up of their metabolism. Endogenous generation of ROS is mediated by way of mitochondrial and non-mitochondrial ROS-generating enzymes which encompass nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox), xanthine oxidase (XO), cytochrome P450 from endoplasmic reticulum (ER), and flavin oxidases from peroxisomes. The number of one resources of ROS generation are the mitochondrial respiration chain and Nox structures.11
To the response of oxidative and nitrosative strain, cells make bigger their antioxidant defenses thru activation of nuclear aspect erythroid 2– (Nrf2)associated problems, an important transcription factor. Nrf2 is a key factor of this manage. structure and recognizes the antioxidant reaction element (ARE) observed within the promoter regions of many genes that encode antioxidants and cleansing enzymes together with heme oxygenase 1 (HO-1), NADPH dehydrogenase quinone 1,Super Oxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1), and Catalase (CAT). Accordingly Nrf2 pathway activation occures to conflict the buildup of ROS and RNS species (reactive nitrogen species). Due to its defensive properties, Nrf2 had been projected as a pharmacological goal in pathologies with neuroinflammatory and oxidative capabilities, along with neurodegenerative and neuropsychiatric diseases. While activated, Nrf2 will increase the expression of countless endogenous antioxidants. And, upon energy irritation and improved ROS levels, as determined at some point of numerous psychiatric episodes, tissue antioxidant defense mechanisms are saturated to the placement they become ineffective.12
Mitochondrial ros production
Mitochondrial electron transport consists of 4 electron reduction of O2 to H2O (Figure 2 ). but, for the duration of mitochondrial electron transport, 1electron reduce in O2 consequences in superoxide
(O2•-). Superoxide anion is detoxified through the mitochondrial manganese superoxide dismutase to yield H2O2, and H2O2 within the presence of decreased transition metals also can be converted to hydroxyl radical (OH-). For the ROS isn’t always originated from mitochondria, peroxisomal β-oxidation of fatty acids turned into considered to be a 2nd supply of oxygen radicals. This reaction generates H2O2 as a by-product. Peroxisomes are organelles accountable for degrading fatty acids with further molecules. Phagocytic cells are every other important source of oxidants, these cells shield the valuable anxious gadget (CNS) in opposition to invading microorganisms and clean the fragments from injured cells with the aid of an oxidative explode of nitric oxide, H2O2 , and O2- .9
The mitochondrion is the number one supply of ROS production in the cells. Below regular physiological condition, up to 2% of the overall cell mitochondrial O2 consumption may additionally be associated to the generation of ROS which includes O2-. Multiple procedures of mitochondrial ROS productions had been proposed, which are regularly modulated by way of the mitochondrial respiration chain complexes. The mitochondrial electron transport chain includes five multi-subunit complexes which includes NADH-coenzyme Q (CoQ) reductase (NADH dehydrogenase, Complex I), succinate dehydrogenase (Complex II), coenzyme Q-cytochrome c reductase (Complex III), cytochrome C oxidase (Complex IV), and ATP synthase (Complex V). Complex I is liable for ROS production of O2- and allows electron transfers from NADH to CoQ. All through this step, protons also are translocated from the matrix to the intermembrane area. Complex II is inside the discount of CoQ and is acknowledged to be worried in generating low degrees of O2- . Complex III, however is concerned technology of O2- in the intermembrane area. The era of O2- is specially greater applicable when the electron transport is reduced with the improved membrane potential. Apparently, the potential of these enzymes to provide ROS may additionally differ number of the organs or during the sickness provisions. For instance, Complex I seems to contribute to the producing of maximum of O2- inside the brain, whilst Complex III is taken in to consideration because the fundamental supply of O2- in the coronary heart and lung . Further, within mitochondrial ETC. Complex I and III are seemed because the most vital manufacturers of O2-. ROS productions from Complex I is around 1/2 of those from complex III in healthy stage, while Complex I exerts the essential position in ROS productions below pathological conditions starting from extended growing to neurodegenerative sicknesses.11 At last, cytochrome P450 enzymes in animals are one of the 1st resistance against herbal poisonous chemical substances from plants. In addition, the era of ROS in dwelling organisms is instently linked with the participation of redox-active metals which include iron and copper. As a standard percept, the chemical beginning place of the majority of ROS is the direct interactions among redox-active metals and oxygen species via reactions inclusive of the fenton and haber-weiss reaction. Free iron (Fe2+ ) reacts thru the Fenton response with H2O2 , leading to the era of very reactive and negative hydroxyl radicals. Superoxide can moreover react with ferric iron within the Haber-Weiss reaction important to the production of Fe2+, which however impacts redox cycling.9 + •O Fe + O2 (Step I) + HO Fe + OH + •OH (Step II) *
Combining step I&II•O2+ HO •OH + HO + O * Known as Fenton reaction.
Apart from direct ROS generation, oblique pathway involves calcium activation with metallo-enzymes together with phospholipases, nitric oxide synthase. Calcium stimulates the tricarboxylic acid cycle and promotes electron glide into the respiratory chain, it also stimulates the nitric oxide synthase and in outcome promotes nitric oxide era, which could inhibit breathing at complex IV.9
Figure 2
Generation of ROS. The superoxide (O2.-) is generated from O2 as a derivative of respiratory chain complex in the mitochondria or by NADPH oxidase. By superoxide dismutase (SOD), the superoxide (O2-) may be converted into hydrogen peroxide (H2O2), which may be in addition transformed to some of other ROS together with hydroxyl radicals (·OH) and hydroxyl anions (OH-).

Excitotoxicity
Glutamatergic neurons appearance the fundamental excitatory system in the brain and play a essential role in lots of neurophysiological capabilities. Underneath normal conditions, glutamate is the predominant neurotransmitter in main perception and cognition within the brain, producing an excitatory response. This reaction is generated following an interplayof glutamate with receptors composing cation channels. Immoderate activation of glutamate receptors can bring about neuronal dysfunction and death, a technique referred to as excitotoxicity. There may be a further of glutamate and glutamatergic responsibility in definite neurodegenerative sickness. The excitatory effects of glutamate are exerted through the use of activation of three fundamental kinds of ionotropic receptors and numerous elegance of metabotropic receptors connected to G-proteins. The fundamental ionotropic receptors activated through glutamate are frequently known as the N-methyl-D-aspartic acid (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) and kainic acid (KA) receptors. These ionotropic receptors are ligand-gated ion channels permeable to exclusive cations.13
Excitotoxicity and ions (NA+, CL -, CA2+)
Acute excitotoxicity is insight to be mediated with the useful resources of excessive depolarization of the postsynaptic membrane. This two results in an osmotic imbalance when countered by means of the use of an inflow of Na+, Cl–, and water, leading to the eventual rupture of cell membranes. Various reviews suggest that acute excitotoxic neurodegeneration following glutamate receptor activation is established on Na+ and Cl– entry. As a consequence, removal of extracellular Na+ or Cl- abolishes NMDA-mediated neurodegeneration.13 Damage of neurons also make contributions to revolutionary long-term neurodegenerative approaches, as an instance the role of L-glutamate (or L-aspartate) mediated acute excitotoxicity in cerebral ischemia or status epilepticus is nicely acknowledged. Microglia-mediated neuroinflammation is moreover one of the maximum placing passage characters of diverse neurodegenerative diseases, such as PD, AD, and ALS.14
Mechanism of excitotoxicity
NMDA receptors are Ca2+ favoring glutamate-gated ion channels that are expressed in most primary neurons and were on the begin held accountable for neuronal damage, closing to their high Ca2+ permeability and conductance characteristics. Continuous activation of big numbers of NMDA receptors (In particularly NR1/NR2B-subtype) leads to growth in intracellular calcium masses and catabolic enzyme activities, which can cause a cascade of events ultimately leading to apoptosis or necrosis. These downstream effects consists of mitochondrial membrane depolarization, caspase activation, production of poisonous oxygen and nitrogen free radicals, and cellular toxicity. The NMDA receptors are additionally grand in mediating excitotoxic neuronal damage. As an example, neurons expressing excessive stages of NMDA receptors are misplaced early inside the striatum of humans affected with neurodegenerative disorders, and injection of NMDA receptor agonists into the striatum of rodents or non-human primates recapitulates the sample of neuronal harm in Huntington’s sickness (HD). AMPA-type glutamate receptors have additionally been implicated in excitotoxicity because assemblies of these receptors are moderately permeable to Ca2+ and in line with chance make contributions to the belated neuronal cell death process brought approximately with the aid of manner of Ca2+ overload. The Ca2+ permeability of the AMPA receptor is decided by the presence or absence of the GluR2 subunit in receptor complex. Low expression of GluR2 allows in the development of AMPA receptors with immoderate Ca2+ permeability and contributes to neuronal degeneration in ischemia. exceedingly, lowering GluR2 tiers or selective blockage of Ca2+ permeable AMPA receptors was additionally tested to guard in opposition to neurodegeneration. However, many more research have examined that demonstrated in glutamate receptor expression after neurological abuse may additionally no longer be so selective.
Consequently, underneath pathological conditions, such as seizures brought about by means of [KA(kainate/kainic acid), a potent agonist of the AMPA/KA sort of glutamate receptors]- or hypoxia-ischemia, many primary cells can also incidence of increase in Ca2+ influx, regardless of the prevailing stoichiometry of AMPA or NMDA receptor assemblies. Numerous latest studies advise that excessive stimulation of non-NMDA glutamate receptors with KA can provoke autophagy and activation of lysosomal enzymes. The autophagy/lysosome pathway in turn, plays an feature in excitotoxic neuronal damage.
In neurodegenerative diseases, metabotropic (mGluR) receptors mediate slow synaptic responses, final to their coupling with intracellular G-proteins. For example, the mGluR1 and mGluR5 subunit subtypes, which exist in huge form of as an alternative spliced forms, are coupled to the inositol trisphosphate (IP3) Ca2+ sign transduction pathway andmay as a consequence affect protein kinase activation and stimulation of Ca2+ release from neuronal stores, both of that could set off behind cell death(Apoptosis) processes. However, mGluR2 also help to mediate the endurance of neurons in the face of selective neuronal disorders and degeneration in AD. Activation of mGluR2 will elevate the phosphorylation of tau and decreases oxidative stress mediated cytotoxicity in neuronal cells. Many appearance of proof shows an extend in glutamate or distinct endogenous glutamatergic agonists in neurodegenerative diseases.
significant proof shows that an excitotoxic response bobbing up from extended extracellular glutamate is probable to be important in locating out the level of tissue damage. An raise in glutamate receptor activity venture induce proapoptotic proteins such as P53, leading to neuronal injury and loss of axistence thru apoptosis and autophagy.13
Protein misfolding and aggregation
The disruption of protein homeostasis (i.e. proteostasis)is that the pathogenic process behind the neurodegenerative diseases at several stage will eventually lead to protein misfolding. Misfolded proteins often cumulative and through neuro cellular stress pathways, gather to generate neurotoxicity and causes the neurodegenerative diseases. Based on the unique brain area that the neurotoxicity affects and usually cause symptoms of neurodegenerative diseases. Neurodegenerative diseases are age related, and their occurrence is predictable to increasing as people keep to stay longer and follow superior survival expectancy.
The common pathology of neurodegeneration consists of deposition of misfolded proteins such as amyloid-β in AD, α-synuclein in PD, TDP-43 in dementia.14
We currently assessment the situation of protein misfolding and aggregation, in accumulation to the position of these phenomena , a few neurodegenerative diseases includes AD, HD, ALS, PD, transmissible spongiform encephalopathies, and spinocerebellar ataxia. By the recognizing of impairing, inhibiting, or reversing protein misfolding as well conferred for the treatment and therapy. Traumatic brain injury (TBI) leads to improved changes of dementia, which includes AD. By the mechanism of trauma can prompt neurodegeneration are an increasing number of implicit. For example, diffuse axonal damage is implicated in disrupting microtubule function, providing the probable context for pathologies of tau and amyloid to develop. The neuropathology of post-traumatic dementias is an increasing number of well characterised, with recent work focusing on chronic traumatic encephalopathy (CTE).15
Chaperones and co-chaperones
The JNK (c-Jun N-terminal kinase) pathway is a central regulator of numerous neuropathologies. Aberrant JNK signalling is implicated in Alzheimer’s, Parkinson’s and Huntington’s diseases, where its activation leads to neuronal cell death. JNK additionally induces neurodegeneration in response to stress stimuli, such as toxins and excitotoxicity increase issue and acute physical injury .Expressed heat shock proteins (Hsp26 and Hsp70) additionally isolated against JNK and Nmnat degeneration phenotypes. These results recommend that molecular chaperones are key in JNK- and Nmnat-regulated axonal defending functions.16
The folding or unfolding of non covalent and the assembly or disassembly of different macromolecular structures are carried by the Molecular chaperones. Chaperones and protein disulfide isomerases (PDI) help nascent peptides keep away from alternative, non-functional, low free potency states .Although many proteins fold into their native states on their own in vitro, chaperones act to stop protein misfolding and possible aggregation . Chaperones are in the main ATP-dependent and characteristic by binding and releasing polypeptides as they fold, mitigating the surface part available for the protein to be negatively influenced by other cellular species. PDIs ensure that disulfide bridges between cysteine residues structure in proper places. In actual representation of their name, chaperone proteins may additionally even get hold of nascent peptide strands as they are translated by using the ribosomes and lead the protein along its subsequent place within the cell. This characteristic is specifically vital to membrane proteins, which should no longer exclusively be inserted into a membrane and fold exact inside it.
Chaperones are recognized to bind numerous proteins under specific conditions, which allow the chaperones to fulfill their function as signaling molecules . When a protein fails to fold properly, even though assist of a chaperone protein, the chaperone will become stranded on the protein. This causes other definite or sequestered proteins to be released, thereby liberating them of their inhibition and allowing them to act as second messengers or activators of packages to make increase the expression of chaperone proteins or to degrade the unwanted proteins . heat shock proteins (HSP) is a usual example for the molecular chaperones, by the way of heat shock a group of proteins are activated. The most outstanding members of this crew are a category of proteins concerned in the folding and unfolding of different proteins. Their expression is amplified when cells are exposed to elevated temperatures, however the Heat shock response(HSR) is additionally brought about at some stage in ischemia, reperfusion, and circulatory and hemorrhagic shock. The well-known motive of HSPs, therefore, is to conflict the denaturing results of these stress-inducing situations; their activation in response to these conditions helps preserve and repair suitable protein folding. chaperonin (i.e. a subclass of molecular chaperone) appeared because the t-complex protein 1 (TCP1) ring complex/chaperonin-containing TCP1(TRIC/CCT). this huge, hetero-oligomeric protein complicated features in an atp-established manner and binds polypeptides internal a imperative hollow space, permitting proteins to fold in an surroundings rather sheltered from the disorderly landscape of the cytosol. tric/cct has a specific set of substrates that it facilitates to fold in eukaryotic cells, collectively with the proteins actin and tubulin, and together with HSP70, this could additionally signify a massive element of the cellular’s protein output. the upsCapabilities in cell well perform by using manner of degrading misfolded proteins, which would otherwise own damaging aspect consequences for cellular characteristic and metabolism. the gadget functions through first ‘tagging’ a rogue protein with more than one ubiquitin molecules and then transporting the tagged protein to the proteasome for degradation. whilst the cellular fails to properly fold a protein and in result fails to harm the protein, these misfolded proteins can gather, subsequentlyCausing the mobile to undergo apoptosis 17
Mechanisms of misfolded protein toxicity
Within the long term, all neurodegenerative disorder proteins produce synaptic disorder and loss ultimately, neuronic dying. the perticular upstream mechanisms by using which distinct misfolded illness proteins cause neurotoxicity are still doubtful, and appear to change counting on the protein species concerned. misfolded illness proteins show up to act specially by way of the usage of toxic gain-offunctionAnd/or dominant-poor outcomes, despite the fact that loss-of-characteristic results have additionally been found. direct, acute outcomes of misfolded proteins on neuronal feature were located after treating neurons with purified oligomers or transfecting them with expression vectors. to provide only some examples amyloid-beta, tau, and alphasynuclein all intrude with synaptic signaling. mutant tau disrupts microtubule feature and neuronal delivery mechanisms and alpha-synuclein disruptsMitochondrial protein import. similarly, advanced aggregates of misfolded proteins may exert poisonous effects by means of binding to and sequestering exclusive cytosolic proteins. The proteomic studies of artificial proteins considered to produce amyloid similar to fibrils showed that the toxicity of these proteins related to the potential of their aggregates to relate in aberrant protein interactions and disrupt the cytosolic trauma reaction. the endogenous cellular proteins are especially sequestered via the amyloid aggregates tended to be extremely big in dimension and enriched in intrinsically unstructured regions, and plenty of play key roles in important cell effects to do together with transcription, translation and protein extremely good control. indeed, some other emerging common function amongst misfolded ailment proteins is their capacity to disrupt proteostasis . Greater these days, cytosolic aggregates of several unique proteins, along with synthetic β-sheets, fragments of mutant huntingtin, and TAR DNA binding protein-43 (TDP-43) have moreover been proven to disrupt nucleocytoplasmic transfer of together proteins and RNA. In increase to synaptic dysfunction, distinct cellular adjustments frequent to the primary neurodegenerative ailments consist of calcium signaling abnormalities, mitochondrial dysfunction, oxidative stress, and neurodegeneration. those symptoms of mobile distress often appear early within the sickness method, and are believed to be a reason as well as a final results of neurodegeneration. this is, the connection between the buildup of misfolded illness proteins and unique symptoms of mobile distress is bidirectional, and generally mutually exacerbating. for example, amyloid-β, a-synuclein, and mhtt all reason acute oxidative stress in neurons and/or astrocytes, and impair astroglial anti-oxidant responses. conversely, oxidative pressure promotes the aggregation of sickness proteins, and contributes to age- and disease-associated proteostatic fall apart. within the equal way, there appear as downward spiralling cycle of interactions between protein misfolding and neuroinflammation, which has been maximum extensively studied for advert. soluble aβ oligomers and insoluble aβ aggregates are proven to bind, prompt, microglia and astrocytes, stimulating a continual low stage situation of neuroinflammation.18
The formation of intracellular neurofibrillary tangles (NFT)I n AD, the tau protein undergoes excessive hyperphosphorylation, essential to the clumping of tau protein. The intracellular formation of NFT leads to microtubule disassembly, dendritic spinal collapse, and the degeneration of axons.19 Several traces of proof suggest that the pro-inflammatory effects of Aβ, even as possibly helpful in the short-term, eventually impair the microglial and astroglial function, including their potential to dispose of Aβ and different misfolded proteins. The neuroinflammation provoked with the relieve of misfolded sickness proteins are probably exacerbated by ongoing, age-related senescence of the immune system.18
Conclusion
At the end this review, it concluded that the neurodegenerative diseases are because of the mechanism of oxidative stress by way of free radicals formation, excitotoxicity, protein missfolding, mitochondrial dysfunction and apoptosis process.